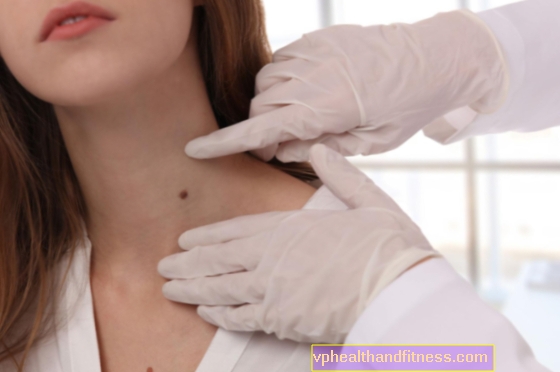
유전병은 드물지만 전 세계 수백만 명에게 영향을 미칩니다. 유럽에서는 130 만에서 260 만 폴란드 인을 포함하여 거의 3 천만 명이 고통을 받고 있습니다. 의학은 일부 유전 질환을 다룰 수 있지만 대부분의 경우에는 무력합니다. 유전병의 원인은 무엇입니까? 그들의 유산은 어떻습니까? 그리고 유전병을 치료할 수 있습니까?
유전 질환은 의학적 발전에도 불구하고 우리가 그것에 대해 거의 알지 못하기 때문에 우려의 원인으로 남아 있습니다. 희귀 (고아) 질병은 10,000 명 중 5 명 미만이 발병하는 질병입니다. 거주자. 그 뿌리에는 유전 적 결함이 있습니다. 이는 신체 (가장 흔히 출생 시부 터)가 적절한 기능에 필요한 물질을 생산하지 않거나 과도하게 축적하지 않음을 의미합니다. 전 세계의 일부 유전 질환은 소수의 사람에게만 영향을 미치고 다른 사람은 수천 명에게만 영향을 미칩니다. 일반적으로 이러한 질병은 어린 시절과 65 %의 경우에 나타납니다. 케이스는 심각합니다. 그들은 종종 개별 신체 기능의 파괴로 이어집니다. 최대 35 % 아픈 아이들은 생후 첫해에 사망합니다. -15 세 이전. 의사들은 따로 분리 된 유전병이 드물게 보이지만 의학적 문제로 볼 때 너무 심각해서 특정한 해결책이 필요하다고 지적합니다. 심각한 경로와 위험한 건강 영향, 그리고 대부분의 경우 치료가 부족하기 때문에 희귀 질환이 유럽 연합에서 공중 보건 활동의 우선 순위 영역으로 확인되었습니다. 요점은 영향을받은 환자가 특정 질병의 희귀 성 및 특정 지역 사회의 경제적 사회적 조건에 관계없이 치료에 동등하게 접근 할 수 있다는 것입니다. 당신은 유전 질환 문제에 관심이 없다고 기꺼이 믿습니다. 때로는 그렇지 않은 것으로 밝혀졌습니다. 따라서 질병이 무엇인지, 누가 위험에 처해 있으며 가족에 나타날 때 도움을 구할 곳을 알아야합니다.
$config[ads_text1] not found
유전병은 어디에서 왔습니까? 유전병의 원인
컴퓨터 과학에서와 같이 비트라고하는 정보 단위가 있으며 유전학에서는 이러한 정보 구성 요소가 유전자이며 유전 정보는 유기체의 구조, 발달 및 기능과 관련이 있습니다. 유전자는 DNA와 단백질로 이루어진 실과 같은 구조에 선형으로 배열되어 있습니다. 염색체. 그들은 그곳에서 영구적 인 위치를 차지하고 엄격하게 정의 된 기능을 수행합니다. 우리는 부모로부터 46 개의 염색체를 물려 받았습니다. 23 개는 어머니로부터, 23 개는 아버지로부터 상속되었습니다 (23 개 염색체의 두 쌍). 1에서 22까지 번호가 매겨진 염색체 (상 염색체)는 남녀 모두 동일하게 보입니다. 쌍 번호 23은 성별 (성 염색체)마다 다릅니다. 성 염색체에는 X와 Y의 두 가지 유형이 있습니다. 여성은 일반적으로 두 개의 X (XX) 염색체를 가지고 있습니다. 하나는 어머니로부터, 다른 하나는 아버지로부터 상속받습니다. 수컷은 어머니로부터 X 염색체를, 아버지 (XY)로부터 Y 염색체를 물려받습니다. 유전 질환은 항상 DNA에 포함 된 유전 정보의 기록 또는 구현 오류의 결과입니다. 하나 이상의 유전자와 관련 될 수 있습니다. 때때로 이들은 염색체의 구조 또는 수의 변화입니다. 하나의 X 염색체 누락 (터너 증후군), 여분의 21 염색체 존재 (다운 증후군)입니다. 유전자의 변화를 염색체에서 돌연변이라고합니다. 대부분의 유전 질환은 여러 유전자의 돌연변이에 의해 조절되는 유전 적 소인으로 인해 발생하는 다 인성 질환입니다. 다양한 환경 요인의 영향을 받아 나타납니다. 여기에는 소위 문명 질병, 예 : 고혈압, 당뇨병, 우울증 또는 정신 분열증 및 소위 고립 된 선천적 결함. 소위 유전 질환 중 희귀 한 단일 대사 질환이 우세합니다.
$config[ads_text2] not found유전병의 전염 위험
정상 또는 변경된 유전자를 상속하는 것은 항상 우리와 완전히 독립적 인 무작위 현상입니다. 때때로 유전 질환이 특정 세대에 나타나지 않습니다. 그러나 때로는 우리에게만 보입니다. 사람이 너무 경미하여 전혀 알아 차리지 못할 때 발생합니다. 또한 자녀가 가족 중 유전 질환의 영향을받는 첫 번째 사람인 경우도 있습니다 (부모 모두가 유전 질환의 보균자가 아님). 이것은 소위에서 난자 또는 정자의 수정에 의해 배아가 형성 될 때 발생할 수 있습니다. 우성 유전자의 새로운 돌연변이는 질병의 증상이 있음을 의미합니다. 그런 다음 부모가 건강하면 다음 자녀도 같은 상태를 갖지 않을 것입니다. 그러나 이미 변경된 유전자를 가진 아이는 자손에게 유전 될 수 있습니다. 환자가 결함이있는 유전자를 물려받을 위험은 50 %입니다. 자녀의 성별에 관계없이. 이것은 상 염색체 우성 유전의 경우입니다. 이러한 방식으로 전염되는 대부분의 질병 (예 : 골격 이형성증, 헌팅턴병)은 새로운 돌연변이의 결과입니다. 우성 유전 질환 중 일부는 출생 시부 터 나타나고 다른 일부는 조금 늦거나 성인기에 나타납니다 (후기 발병 질환). 희귀 유전 질환 중 상 염색체 열성 유전이있는 대사 질환이 우세합니다. 열성 유전 (예 : 폼 페병, 고셔병)의 경우, 질병의 증상은 아이가 부모 (엄마와 아빠에게서 하나씩)로부터 동일한 유전자의 두 개의 변경된 사본을받을 때만 나타납니다. 다음 자손에게 질병이 재발 할 위험은 25 %입니다. 이후 임신 할 때마다 소년과 소녀 모두 일정합니다. 아이가 하나의 나쁜 유전자와 좋은 하나의 유전자를 물려 받으면 (50 % 확률) 부모처럼 건강한 보균자가 될 것입니다. 아이가 좋은 유전자 사본을 모두 물려 받으면 질병이나 보균자의 영향을받지 않습니다.
$config[ads_text3] not found 도움을받을 수있는 곳
도움이 필요하십니까?
- 희귀 질환, 임상 시험, 의약품, 유럽 전역의 협회 및 지원 그룹에 대한 온라인 정보 서비스,
- 희귀 질환 치료를위한 고아 국가 포럼,
- 파브리 병 가족 협회,
- Mucopolysaccharidosis 및 관련 질병을 가진 환자의 연관성,
- 폴란드 인간 유전 학회,
- 의사, 환자 및 부모를위한 유전 질환에 대한 정보를 제공하는 유전학 및 생명 공학 포털입니다.
딸이 유전병을 물려받을 때와 아들이
X- 연관 유전 질환은 해당 염색체의 유전자 변화로 인해 발생합니다. 남성은 X 염색체와 관련된 열성 질환 (예 : 혈우병, 점액 다당류 증 II 형)으로 고통받는 반면 여성은 유전자 돌연변이의 운반자입니다. 어머니가 특정 질병의 유전자에 돌연변이를 가지고 있다면, 그녀가 아들에게 잘못된 유전자를 가진 X 염색체를 물려 줄 확률은 50 %입니다. 임신 횟수에 관계없이 각 딸의 경우, 어머니로부터 결함 유전자를 물려받을 위험도 50 %입니다. 딸이 잘못된 유전자를 물려 받았을 때, 마치 어머니가 질병 유전자 돌연변이의 건강한 운반자가 될 것입니다. 남자는 항상 변경된 유전자를 딸에게 물려 주므로 모든 딸은 보균자가 될 것입니다. 우세한 특징으로 유전되는 X- 연관 질환은 매우 드뭅니다. 돌연변이를 가진 여성은 질병의 매우 경미한 증상을 보일 수 있습니다. 남성과 여성의 자손에게 돌연변이를 전파 할 확률은 50 %입니다. 그러나 소년의 경우 질병의 진행이 매우 심할 수 있으며 때로는 임신이 저절로 죽습니다. 여아의 경우 증상은 일반적으로 경미합니다.
$config[ads_text4] not found유전 상담이 진단을 내립니다
때때로 소아는 유전 질환의 증상을 가지고 태어나지 만 종종 심각한 유전 질환이 점차적으로 발생하며 첫 번째 징후는 무관심, 이동성 감소, 수유 장애 및 느린 체중 증가입니다. 소아과 적으로 정당화되지 않는 아동의 발달이나 행동에 관계없이 모든 방해 신호에는 유전 적 상담이 필요합니다. 폴란드의 모든 의료 아카데미에는 유전 상담 센터 또는 분자 유전학 부서가 있습니다.전문 검사 결과에 따라 유전 의사는 진단을 내릴뿐만 아니라 소위 유전 상담. 특히, 유전 위험 평가, 즉 질병이 다른 어린이에게서 재발 할 가능성에 대한 질문에 답합니다. 어떤 경우에는 질병을 진단하는 데 한 달이 걸리고 다른 경우에는 훨씬 더 오래 걸립니다. 예를 들어, I 형 점액 다당류 증은 종종 아이가 1 세가되기 전에 진단됩니다. 성인의 진단에서는 더 나쁩니다. 한 독일인은 첫 징후가 나타난 지 44 년 만에 파브리 병 진단을 받았습니다. 그 과정에서 그는 다양한 질병, 심지어 정신과 치료를 받았습니다. 그러나 그것은 진단하기 매우 어려운 질병입니다. 전문가의 진단을 돕기 위해 희귀 사례 보고서 수집 센터가 전 세계적으로 만들어지고 있습니다. 의사는 항상 면담 (질병의 증상에 대한 질문)으로 시작하여 건강 검진을 실시하고 혈통 (가족의 유전 질환에 대한 정보)을 분석합니다. 유전병이 의심되면 유전자 검사를 권합니다.
유전 연구의 유형
보편적 인 유전자 검사는 없으며 그 유형은 질병의 유형, 유전 적 배경에 따라 달라지며 건강 보고서 (예 : 혈우병, 점액 다당류 증 II 형)는 남성에게 영향을 미치고 여성은 유전자 돌연변이의 보균자라는 사실을 아는 것이 중요합니다. 어머니가 특정 질병의 유전자에 돌연변이를 가지고 있다면, 그녀가 아들에게 잘못된 유전자를 가진 X 염색체를 물려 줄 확률은 50 %입니다. 임신 횟수에 관계없이 각 딸의 경우, 어머니로부터 결함 유전자를 물려받을 위험도 50 %입니다. 딸이 잘못된 유전자를 물려 받았을 때, 마치 어머니가 질병 유전자 돌연변이의 건강한 운반자가 될 것입니다. 남자는 항상 변경된 유전자를 딸에게 물려 주므로 모든 딸은 보균자가 될 것입니다. 우세한 특징으로 유전되는 X- 연관 질환은 매우 드뭅니다. 돌연변이를 가진 여성은 질병의 매우 경미한 증상을 보일 수 있습니다. 남성과 여성의 자손에게 돌연변이를 전파 할 확률은 50 %입니다. 그러나 소년의 경우 질병의 진행이 매우 심할 수 있으며 때로는 임신이 저절로 죽습니다. 여아의 경우 증상은 일반적으로 경미합니다. 과학자들은 우리의 DNA에 잘못된 기록으로 인한 질병을 통제하는 데 도움이 될 효과적인 유전자 치료법을 끊임없이 찾고 있습니다. 현재로서는 이러한 질병의 일부 증상을 완화 할 수 있습니다. 다릅니다. 따라서 질병이 무엇인지, 질병의 의심을 확인하는 검사를 수행하는 기술적으로 누가 가능한지 알아야합니다. 이것은 핵형 검사, 특정 효소 또는 단백질 생성물의 활성, 경우에 따라 유전자 돌연변이를 찾기위한 DNA 분석 일 수 있습니다. 테스트 재료는 일반적으로 혈액 샘플입니다. 폴란드 나 세계에 모든 유전자 검사가 가능한 실험실은 없습니다. 특정 질병이 드물기 때문에 모든 국가에서 특정 연구를 수행하는 것도 의미가 없습니다. 따라서 유럽의 일부 센터는 매우 희귀 한 질병의 진단을 전문으로합니다.
유전 질환 발병 위험을 결정하기위한 임신 전 또는 임신 중 검사
유전병은 예방할 수 없습니다. 따라서 그러한 질병이 가족에서 발생했거나 가족 또는 파트너 중 누군가가 특정 질병의 돌연변이를 가지고 있다면 가정의와 상담하는 것이 좋습니다. 그런 다음 상담을 위해 유전 의사에게 의뢰해야합니다. 그곳에서 당신은 당신의 두려움이 정당한지, 그리고 있다면 당신과 당신의 가장 가까운 친척들에게 어떤 검사를해야하는지 알게 될 것입니다. 임신 중에 일부 유전 적 상태를 검사하여 아기의 건강을 확인할 수 있습니다 (산전 선별 검사). 침습적 산전 검사는 소위 유전 적 위험군, 즉 특정 질병의 확률이 높은 것으로 이전에 알려진 가족. 이러한 유형의 진단에 대한 적응증은 일반적으로 임상 유전학자가 결정합니다.
특정 희귀 유전 질환
폼 페병-근육 손상은 세포에서 자연적으로 발생하는 효소의 결핍 또는 오작동으로 인해 발생합니다. 생후 첫 달에 나타날 수 있으며 가장 위험합니다. 일반적으로 혈액 순환이나 호흡의 합병증으로 인해 생후 1 년 이내에 사망합니다. 후기 발병 환자는 다리 근육의 점진적인 약화, 호흡 문제 등 다양한 심각도의 증상을 보입니다. 목발이나 수레의 도움으로 움직이며 호흡 보조가 필요합니다.
Maroteaux-Lama 질병 (mucopolysaccharidosis VI)-점액 다당류 분해를 담당하는 효소가 부족합니다. 완전히 분해되지 않은 물질은 뼈와 연골의 적절한 발달에 사용할 수 없으며 신체의 세포에 축적되어 점진적으로 파괴됩니다.
Fabry 질병-유전자 기록의 오류로 인해 신체에 a-GAL 효소가 부족하게되는데, 그 임무는 세포가이를 재사용하거나 배설하기 위해 특정 물질을 분해하는 것입니다. 결과적으로 이러한 물질은 혈관 벽을 구성하는 세포에 축적됩니다. 치료하지 않고 방치하면 신체의 주요 시스템이 손상되고 사망에 이르게됩니다.
Gaucher 질병-그 본질은 신체의 특정 지방 물질을 분해하는 효소의 결핍 또는 부족입니다. 결과적으로 신체의 다른 부분에 침착됩니다. 질병을 치료하지 않으면 뇌, 간, 비장, 뼈, 혈액과 같은 다른 기관의 손상으로 인해 심각한 장애가 발생하여 결과적으로 사망에 이르게됩니다.
일부 유전 질환은 치료할 수 있습니다
아이가 유전 적 조건을 가지고 있다는 사실에 항상 놀라게됩니다. 분노, 충격, 절망은이 상황에서 모두 정상적인 반응입니다. 많은 문제를 처리해야 할뿐만 아니라 "나쁜"유전자를 자녀에게 전달했다는 죄책감에도 시달립니다. 당신은 정말로 그것을 통제 할 수 없다고 믿어야합니다. 의료 및 심리 치료에도 불구하고 아픈 자녀의 부모는 종종 무력감과 소외감을 느낍니다. 사람들은 일반적으로 불행에 대처하는 방법을 모르기 때문에 그러한 가족과 거리를두기 때문입니다. 따라서 같은 운명을 가진 가족의 지원을 찾으십시오. 세계에서 그리고 점점 더 자주 우리나라에서는 다양한 유전 질환의 영향을받는 가족 협회가 형성됩니다. 불행히도 유전병은 치료할 수 없습니다. 변경된 유전자는 복구 할 수 없으며 평생 동안 존재합니다. 종종 의사의 역할은 고통받는 환자를 동반하는 것입니다. 그러나 최근 몇 년 동안, 최근까지 치명적이라고 여겨지 던 일부 유전 질환은 치료가 가능하다는 것이 분명해졌습니다. 즉, 감소하거나 제거 할 수도 있습니다. 시작된 치료는 평생 동안 지속되어야합니다. 왜냐하면 치료를 중단하면 증상이 두 배의 힘으로 회복되기 때문입니다. 이 질병은 과학자들이 효과적인 유전자 치료법을 발견 할 때까지 여전히 인간에게있을 것입니다. 인과 적 치료는 페닐 케톤뇨증에 대한 최초의 치료였으며 배설식이 요법을 기반으로합니다. 일부 희귀 대사 질환은 이제 신체가 생산할 수없는 분리 된 효소를 정맥 내 주입하는 대체 요법으로 치료됩니다. 효소 대체 치료는 대사 질환 병동의 바르샤바에있는 어린이 건강 센터에서 실시합니다. 비용이 매우 비쌉니다 (예 : 폼 페병 환자 18 명의 연간 치료 비용은 약 740 만 PLN입니다). 일부 환자의 치료는 자선을 위해 회사에서 자금을 지원합니다. 불행히도 대부분의 유전병에 대한 치료법은 없습니다.
월간 "Zdrowie"